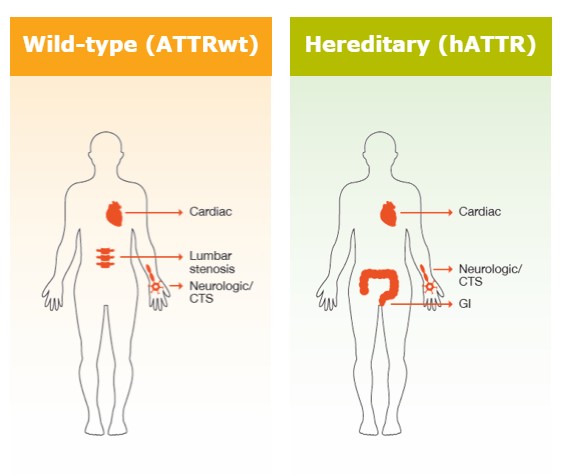
Il existe deux formes d’ATTR-CM : l’ATTR-CM de type sauvage et l’ATTR-CM héréditaire8. Chacune a une cause différente.
L'ATTR-CM de type sauvage (wtATTR-CM) survient principalement chez les patients âgés de plus de 60 ans ; c'est pourquoi elle est également appelée "amylose cardiaque liée à l'âge". Les hommes sont plus sensibles que les femmes. La cause de cette maladie n'a pas encore été élucidée3,9,10.
En outre, il existe une forme héréditaire d'ATTR-CM (hATTR-CM). Les patients atteints d'hATTR peuvent être relativement jeunes lorsque les premiers symptômes apparaissent, et l'évolution de la maladie peut varier considérablement d'un patient à l'autre. Cette forme est rare et touche principalement les hommes9,21.
Contrairement au type sauvage, la cause de l'ATTR-CM héréditaire est bien connue. En effet, les patients atteints d'hATTR-CM présentent certaines modifications dans leur matériel génétique, connus sous le nom de « mutations génétiques ». Ces mutations peuvent conduire à des formes moins stables de la protéine transthyrétine. Mondialement, plus de 100 mutations sont connues pour être associées à l'ATTR héréditaire. En fonction du type de mutation, l’affection prendra une évolution spécifique9,21.
Comme les parents peuvent transmettre les mutations génétiques à leur progéniture, on parle de forme héréditaire, "hereditary" en anglais, d'où le "h" dans la désignation hATTR-CM9.

À PROPOS DE L’ATTR-CM
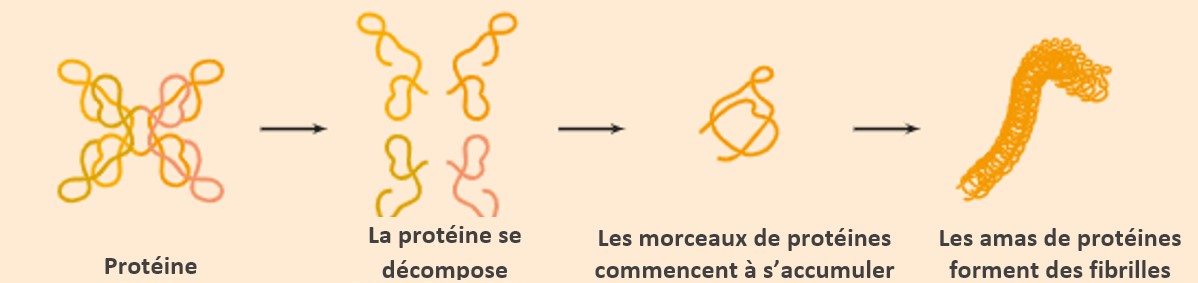
Notre corps fabrique constamment des protéines. Elles sont les éléments constitutifs de toutes les cellules. Ces protéines se replient dans une certaine forme.
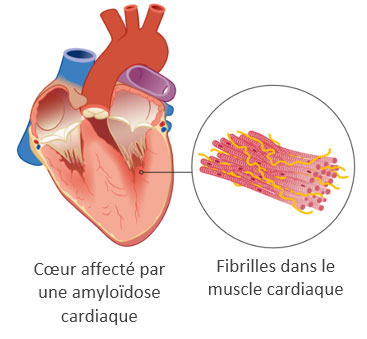
Dans l'amylose ATTR - nom complet : amylose à transthyrétine - il y a un problème dans la façon dont la protéine transthyrétine est repliée. C'est pourquoi la maladie est également appelée "maladie du repliement des protéines". En raison du mauvais repliement, des protéines « instables » se forment, qui s’agglutinent. Les chaînes de protéines ainsi formées sont appelées « fibrilles amyloïdes »1,2. L'organisme ne pouvant plus traiter ou excréter ces fibrilles, celles-ci se déposent dans les organes ou les tissus et y provoquent des lésions.
Selon l'endroit où les fibrilles se déposent dans l'organisme, la maladie peut se manifester de différentes manières, par exemple, au niveau du cœur, ces fibrilles entraînent des troubles cardiaques, au niveau des reins, une insuffisance rénale, au niveau du système nerveux, des douleurs chroniques ou des troubles sensoriels.3,4
Sans traitement, les patients atteints d'amylose ATTR ont une espérance de vie moyenne de 3 à 5 ans à partir du moment du diagnostic. Avec un traitement approprié, il est possible de ralentir le processus de la maladie et d'allonger l'espérance de vie.5
ATTR-CM signifie cardiomyopathie (CM) amyloïde à transthyrétine. 6,7 En cas d’ATTR-CM, les fibrilles amyloïdes se déposent dans les ventricules ou cavités cardiaques. Ce sont les cavités dans le cœur grâce auxquelles, par contraction du muscle cardiaque, le sang est envoyé dans les artères.
Par le dépôt des fibrilles amyloïdes, les muscles des ventricules cardiaques deviennent plus épais et se rigidifient. Cela finit par réduire l'action de pompage du cœur, empêchant le sang riche en oxygène d'être pompé de manière adéquate et provoquant des maladies cardiaques graves.8
Lorsque l'ATTR-CM est suspectée sur la base de certains symptômes, des tests spécialisés peuvent confirmer la maladie. Les principaux tests sont l'échocardiographie (ECG), complétée par des techniques d'imagerie avancées (telles que l'IRM et la scintigraphie osseuse), des tests de laboratoire et des tests génétiques10,11.
Chez les patients souffrant de la forme héréditaire, il peut être utile de tester également les membres de la famille proche. En effet, les membres de la famille dont le test est positif peuvent être suivis de près et recevoir un traitement approprié dès l'apparition des symptômes9.
Même après le diagnostic, il est important que les patients se soumettent à des examens réguliers - notamment des échocardiographies et des analyses de sang - pour surveiller l'évolution de la maladie.
Votre médecin décidera des examens utiles pour vous et vous en informera.
La progression de la maladie varie d’un patient à l’autre et dépend de l'état général, de l'âge et, surtout, du stade auquel la maladie est diagnostiquée.
Un diagnostic précoce est important pour pouvoir stabiliser au mieux la maladie et retarder son évolution. En effet, une fois le diagnostic posé, il est possible de commencer un traitement et de ralentir l'augmentation des fibrilles amyloïdes dans l'organisme. Cela améliore la qualité de vie et prolonge l'espérance de vie.
Mais comme les patients peuvent présenter des symptômes très divers, à la fois cardiaques et non cardiaques, des symptômes qui ne sont pas non plus très spécifiques, il n'est pas toujours facile de poser un diagnostic à un stade précoce. En cas de diagnostic tardif, le pronostic est moins favorable.
Les symptômes dépendent de l’endroit du corps où se dépose l’amyloïde. L’ATTR-CM étant une maladie du muscle cardiaque, elle entraîne souvent des problèmes cardiaques. Les symptômes sont très similaires à ceux de l’insuffisance cardiaque, mais peuvent varier d’un patient à l’autre.14-16
Symptômes | Plaintes |
|---|---|
Insuffisance cardiaque | • Essoufflement (dyspnée) |
Les autres caractéristiques connexes et communes fréquentes sont les suivantes : 2, 16, 17
Symptômes | Plaintes |
|---|---|
Polyneuropathie | • Troubles sensoriels/douleur à différents endroits du corps par atteinte du système nerveux périphérique |
Syndrome du canal carpien (bilatéral) | • Sensation sourde, de type picotements ou douloureuse dans la main et les doigts due à un pincement du nerf au niveau du(des) poignet(s) |
Sténose spinale | • Jambes douloureuses et fatiguées (sensation de lourdeur) |
Outre le cœur, le système nerveux peut également être affecté, entraînant une polyneuropathie (trouble du système nerveux périphérique).
Le syndrome du canal carpien, ou compression du nerf dans le canal du poignet, survient chez certains patients à un stade précoce de la progression de la maladie, avant tout autre symptôme détectable. Ce syndrome peut être un signe précoce de l’ATTR-CM, mais pas nécessairement toujours.
Comme le tableau clinique peut varier considérablement d’un patient à l'autre, il est possible que d’autres symptômes non cardiaques supplémentaires apparaissent : 16, 18
Symptômes | Plaintes |
|---|---|
Symptômes gastro-intestinaux | • Diarrhée |
Intolérance aux médicaments contre l’insuffisance cardiaque11, 17, 19 | - |
Rupture du tendon du biceps | - |
Problèmes de la hanche ou du genou pouvant conduire à une prothèse18 | - |
Affections oculaires4 | • Pression oculaire |
Compte tenu du manque de spécificité de ces plaintes, un diagnostic correct n’est pas toujours évident à poser.3,4
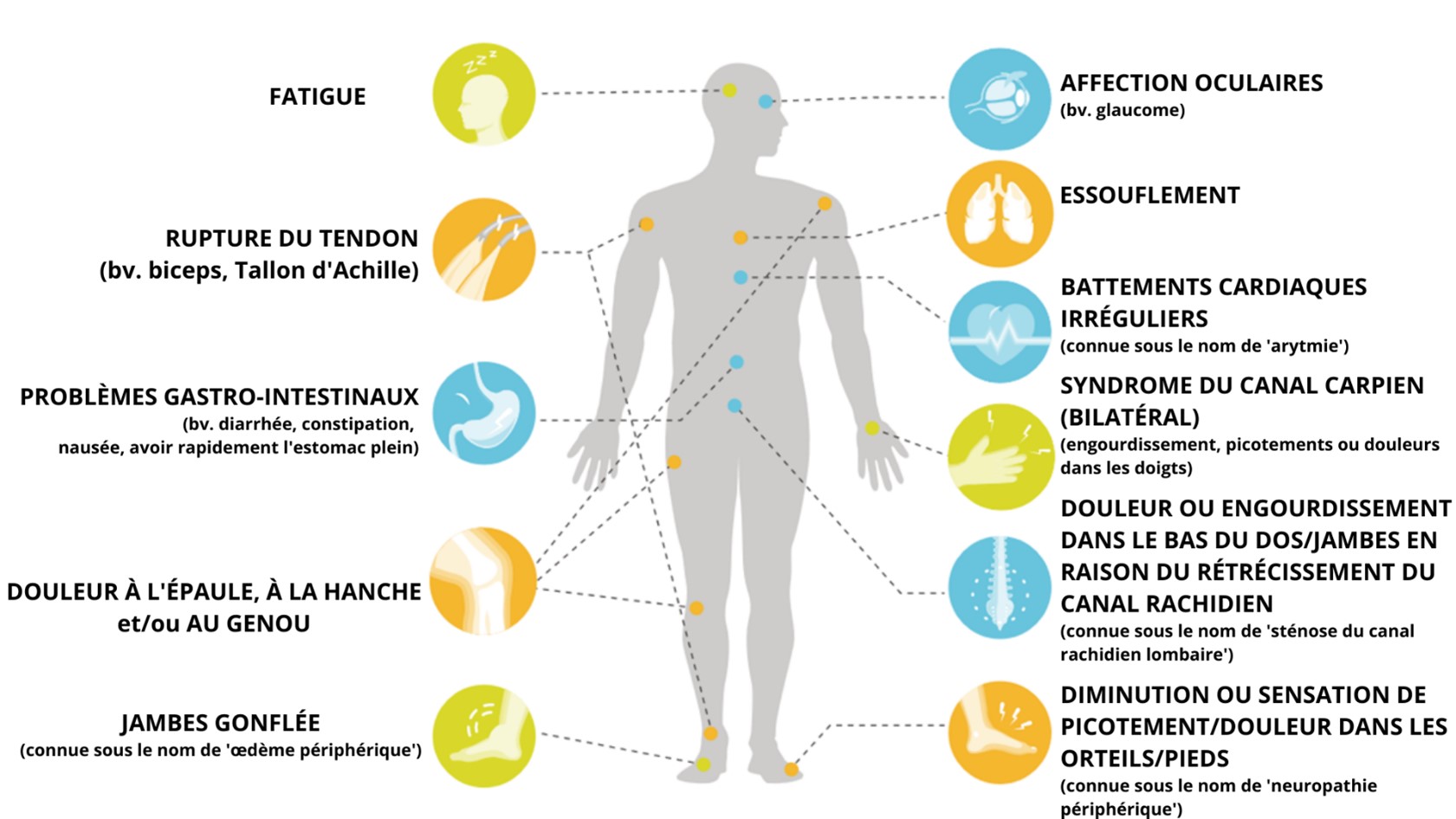
Ces exemples sont à titre indicatif seulement. Les symptômes et les plaintes peuvent varier d'un patient à l'autre.
L'amylose cardiaque n'est pas guérissable aujourd'hui. Cependant, la maladie est de plus en plus traitable grâce à des médicaments qui peuvent ralentir le processus de la maladie4. Votre médecin pourra vous en dire plus.
En fonction des symptômes spécifiques que vous présentez, votre médecin pourra également vous prescrire des médicaments pour diminuer les symptômes gênants. Il peut s'agir de médicaments diurétiques, par exemple, ou de médicaments pour traiter l'insuffisance cardiaque ; les douleurs éventuelles peuvent être contrôlées à l'aide d'analgésiques.
Comme c'est le foie qui produit la protéine instable transthyrétine, une greffe de foie peut être envisagée dans certains cas. Cependant, cette procédure n'est pas possible pour tout le monde et comporte des risques. Si vous remplissez les conditions requises, votre médecin vous en informera certainement.
Enfin, soyez également attentif à votre mode de vie. Une alimentation adaptée et une activité physique suffisante, par exemple, peuvent avoir un impact positif sur votre qualité de vie générale. Demandez conseil à votre médecin à ce sujet.13,20
1.Siddiqi OK, Ruberg FL. Trends Cardiovasc Med. 2018;28(1):10–21.
2.Halwani O, Delgado DH. Expert Rev Cardiovasc Ther. 2010;8(7):1007–1113.
3.Donnelly JP, Hanna M. Cardiac amyloidosis: an update on diagnosis and treatment. Cleve Clin J Med. 2017;84(12 suppl 3):12-26.
4.Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, Gospodinova M, Obici L, Rapezzi C, Garcia-Pavia P. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019 Aug;7(8):709-716. doi: 10.1016/j.jchf.2019.04.010. Epub 2019 Jul 10. PMID: 31302046.
5.Ruberg, F.L., Grogan, M., Hanna, M., Kelly, J.W., Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol, 2019, 73 (22), 2872-2891.
6.González-López E, López-Sainz Á, Garcia-Pavia P. Diagnosis and treatment of transthyretin cardiac amyloidosis. Progress and hope. Rev Esp Cardiol. 2017;70(11):991-1004.
7.Quarta CC, Solomon D, Uraizee I et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129(18):1840-1849.
8.Maurer MS, Hanna M, Grogan M et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol,. 2016;68(2):161-172.
9.Oerlemans MIFJ, Rutten KHG, Minnema MC, Raymakers RAP, Asselbergs FW, de Jonge N. Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 2019;27(11):525-536.
10.Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.
11. Narotsky D, et al. Wild-type transthyretin cardiac amyloidosis: novel insights from advanced imaging. Can J Cardiol. 2016;32(9):1166.e1-1166.e10
12. TTR-FAP: A Quick Guide for General Practitioners. Pfizer 2017.
13. Adams D. et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016, 29 (suppl 1):S14–S26.
14. Mayo Clinic: Heart failure. http://www.mayoclinic.org/diseases-conditions/heart-failure/symptoms-causes/syc-20373142. Accessed May 8, 2019.
15. Ponikowski P, et al. Eur Heart J. 2016;37(27):2129–2200.
16. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, Grogan M, Kristen AV, Lousada I, Nativi-Nicolau J, Cristina Quarta C, Rapezzi C, Ruberg FL, Witteles R, Merlini G. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019 Sep;12(9):e006075. doi: 10.1161/CIRCHEARTFAILURE.119.006075. Epub 2019 Sep 4. PMID: 31480867; PMCID: PMC6736650.
17. Brunjes DL, Castano A, Clemons A, Rubin J, Maurer MS. Transthyretin cardiac amyloidosis in older Americans. J Card Fail. 2016;22(12):996–1003.
18. Rubin J, Alvarez J, Teruya S, Castano A, Lehman RA, Weidenbaum M, Geller JA, Helmke S, Maurer MS. Hip and knee arthroplasty are common among patients with transthyretin cardiac amyloidosis, occurring years before cardiac amyloid diagnosis: can we identify affected patients earlier? Amyloid. 2017 Dec;24(4):226-230. doi: 10.1080/13506129.2017.1375908. Epub 2017 Sep 14. PMID: 28906148.
19. Castaño A, Drach BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-178.
20. Pinney JH & Hawkins PN. Amyloidosis. Review Article. Ann Clin Biochem 2012; 49: 229–241. DOI: 10.1258/acb.2011.011225.
21. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016;68(12):1323-41


