
À PROPOS DE L’ATTR-CM
L’ amylose ou amyloïdose est une maladie qui se caractérise par l’accumulation de dépôts de protéines, des molécules fabriquées en permanence par l’organisme, qui se replient dans une forme incorrecte. Elles s’agglutinent pour former des chaînes de protéines insolubles : les fibrilles amyloïdes.
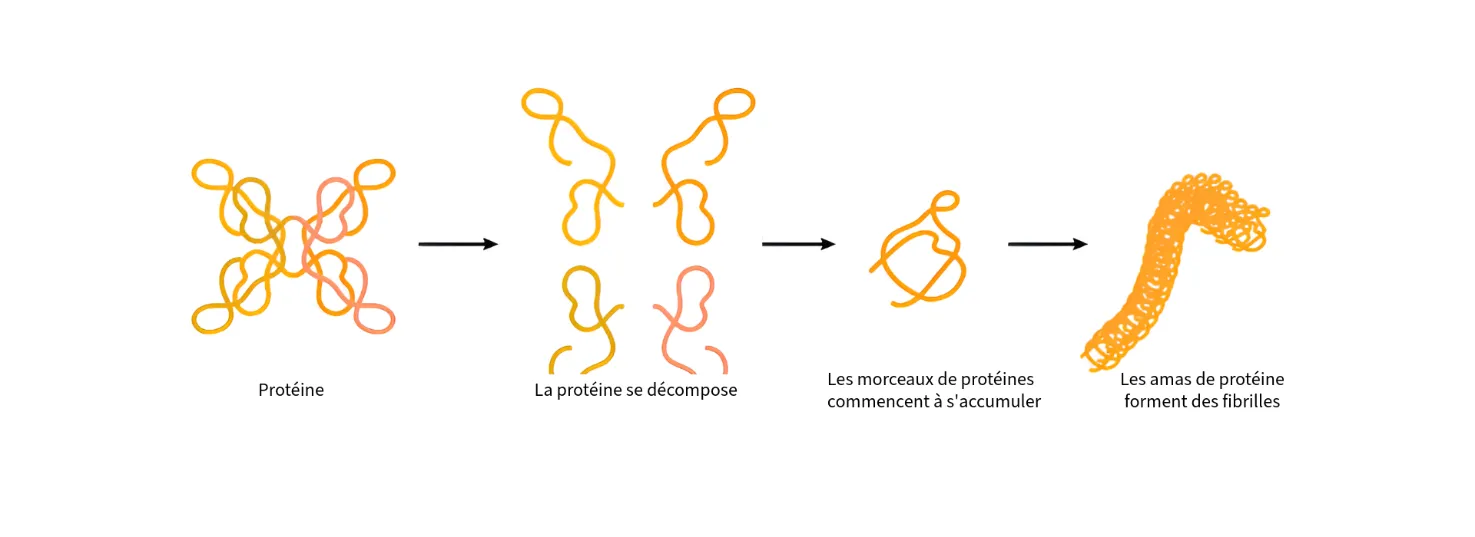
L’ organisme ne parvient plus à éliminer les fibrilles, qui s’accumulent dans les organes et tissus, provoquant des lésions. Selon leur localisation, la maladie se manifeste différemment : atteinte cardiaque, insuffisance rénale ou troubles neurologiques (douleurs, troubles sensoriels, etc.).
L’amylose TTR
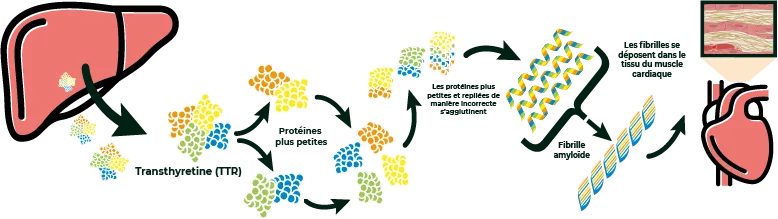
L' amylose à transthyrétine est une forme spécifique d’amylose associée à la protéine transthyrétine. Cette protéine, qui circule dans le sang à l’état normal, est responsable du transport des hormones throïdiennes et du rétinol. D’où son nom : TRANS-THY-RÉTINE, ou TTR en abrégé. 1
2 formes d’amylose TTR :
Amylose acquise (TTR de type sauvage)
Amylose héréditaire
Cette forme rare touche autant les femmes que les hommes, avec des symptômes apparaissant généralement entre 50 et 60 ans.
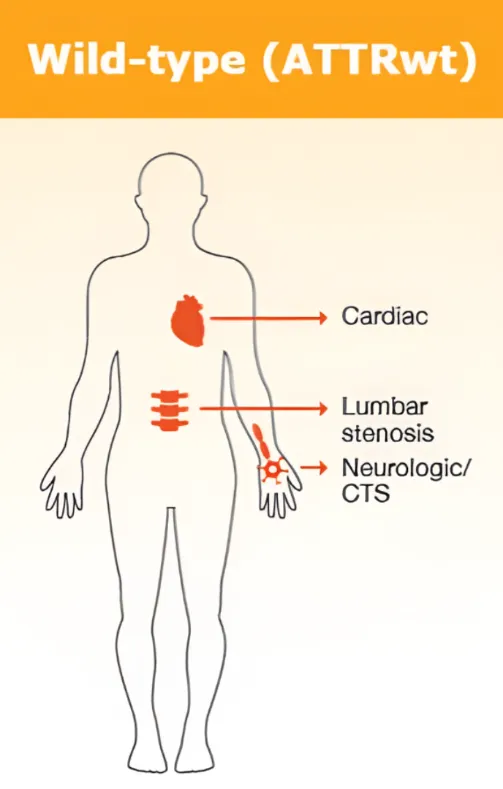
Amylose acquise (TTR de type sauvage) 1
La forme la plus fréquente d’amylose TTR est l’amylose acquise, également appelée amylose à TTR de type sauvage. Cette forme est liée à l’âge et touche principalement les hommes de plus de 60 ans. La fréquence de la maladie augmente en fonction de l’âge, raison pour laquelle on parle aussi d’amylose liée à l’âge voire d’amylose sénile. Les femmes peuvent également développer la maladie, mais généralement à un âge beaucoup plus avancé.
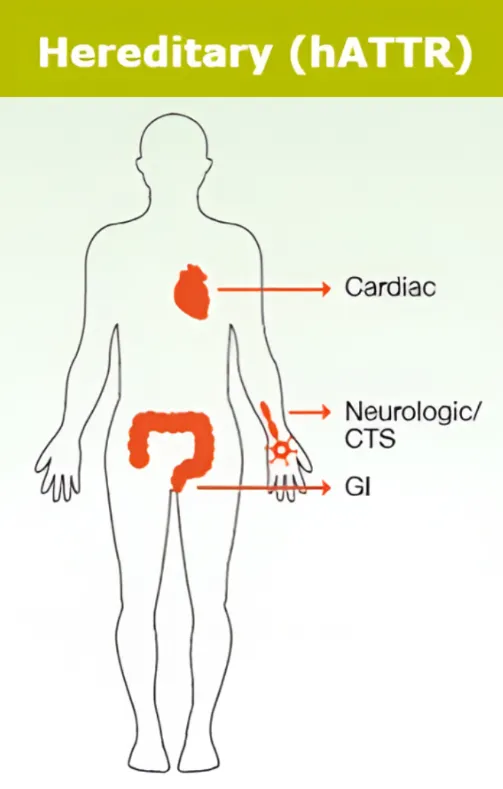
Forme héréditaire de l’amylose TTR 1,2
Cette forme rare touche autant les femmes que les hommes, avec des symptômes apparaissant généralement entre 50 et 60 ans. Elle est liée à des mutations génétiques affectant la stabilité de la transthyrétine. Plus de 120 mutations sont connues, chacune influençant l’évolution de la maladie. La transmission étant héréditaire, un test génétique est recommandé pour les proches. En cas de résultat positif, un suivi précoce et un traitement rapide sont essentiels, car cette forme peut évoluer plus vite que la forme acquise.
C’EST UNE MALADIE HÉRÉDITAIRE ... ET VOS ENFANTS ? 5
L’amylose ATTR pouvant être une maladie héréditaire, cela signifie que si vous ou votre partenaire est porteur d'une mutation du gène qui code pour la transthyrétine, vos enfants ont 50 % de chances d‘avoir la maladie également. S’ils n’ont pas le gène malade, ils ne peuvent donc pas le transmettre à leurs enfants.
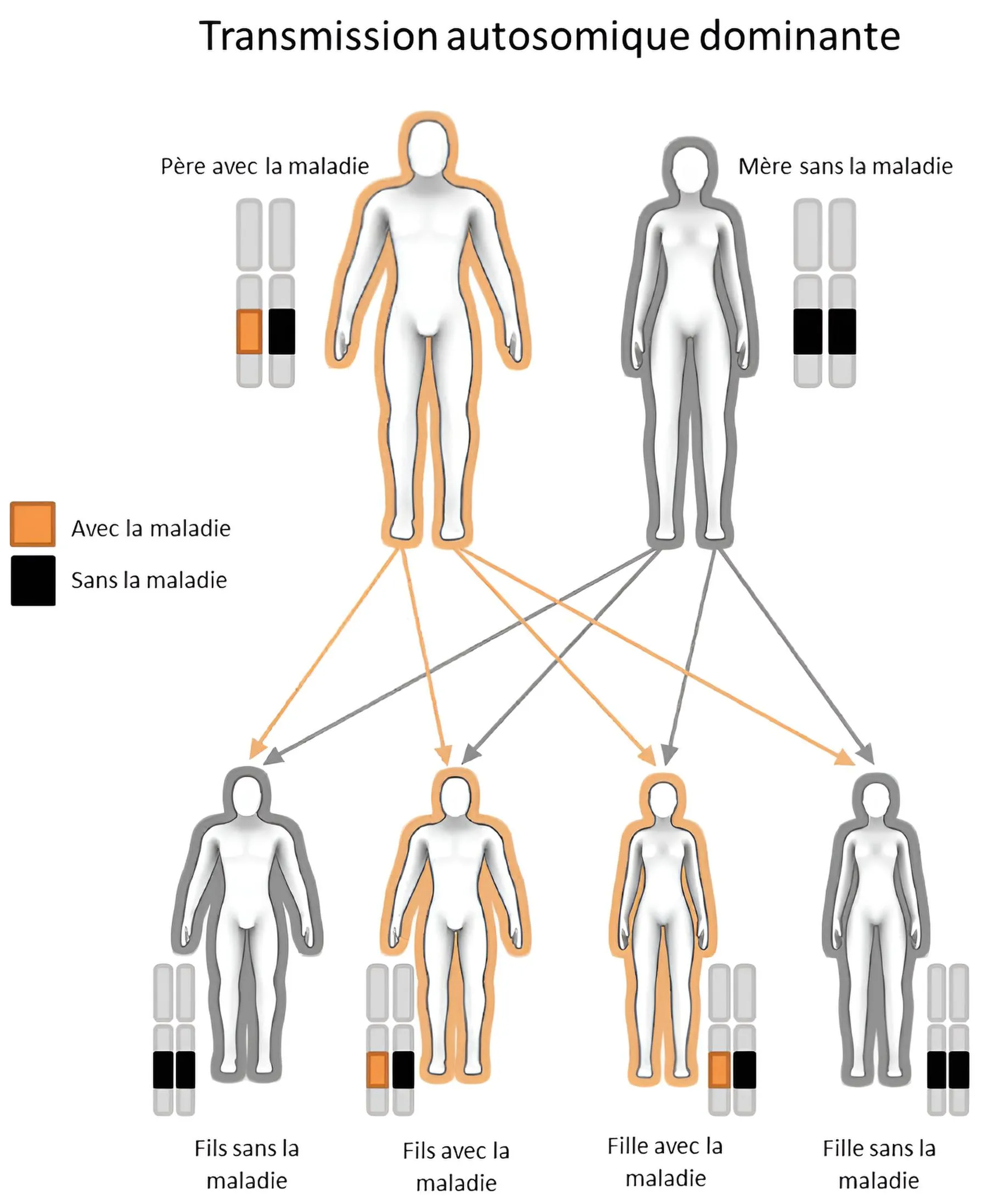
Parce qu’il s’agit d’une maladie héréditaire, il est utile de vérifier si un membre de votre famille présente ou a présenté des symptômes similaires dans le passé. N’oubliez pas de le signaler à votre médecin !
Un diagnostic précis est essentiel pour choisir un traitement adéquat
En cas de suspicion d’amylose, le médecin demandera une série de tests pour confirmer la présence de fibrilles amyloïdes.
Des tests complémentaires sont nécessaires pour déterminer le type d’amylose et les organes affectés par les fibrilles (par exemple, le coeur, le foie, les reins ou les nerfs).
QUEL EXAMEN ? | QUEL EN EST L’OBJECTIF ? |
|---|---|
Electrocardiographie | Ils permettent de mesurer la fonction cardiaque et d’en rechercher toute modification (troubles du rythme et de la conduction). |
Échocardiographie | Ces examens permettent d’étudier l’implication du muscle cardiaque (le myocarde), des valves cardiaques et des vaisseaux sanguins qui transportent l’oxygène vers le muscle cardiaque (artères coronaires). |
Electro(neuro)-myographie (EMG) | Cet examen permet de vérifier les modifications de la fonction nerveuse dues au dépôt de fibrilles amyloïdes (trouble neurologique). |
Scintigraphie osseuse | Ce test permet de vérifier si le muscle cardiaque est affecté par les fibrilles amyloïdes et d’estimer les dépôts dans l’organisme. Par rapport à une biopsie, ce test ne nécessite pas d’intervention chirurgicale mineure pour prélever du tissu, raison pour laquelle il est de plus en plus souvent choisi. |
Examen histologique | Sous anesthésie locale, un petit échantillon du muscle cardiaque ou d’un autre organe, comme la graisse abdominale, est prélevé (biopsie). Au microscope, on examine ensuite si des fibrilles amyloïdes sont présentes dans le tissu. |
Détermination des chaînes légères libres | Ce test est effectué pour exclure l’ amylose à chaînes légères (Amylose AL). Dans un échantillon de sang ou d’urine, on vérifie la quantité de protéines à chaînes légères libres (ou non liées) dans votre organisme. Si la quantité de ces protéines libres est supérieure à la normale, cela peut signifier que vous êtes atteint(e) d’amylose AL ou d’une autre maladie. |
Test génétique | Pour confirmer ou infirmer une forme héréditaire de l’amylose, l’équipe soignante vous fera également passer un test génétique (par le biais d’une analyse de sang).
|
Note: Même après l’établissement du diagnostic, un certain nombre d’examens seront effectués régulièrement pour vérifier l’évolution de la maladie.
Dans l’état actuel des choses, l’amylose TTR cardiaque n’est pas guérissable, mais peut être ralentie. Il existe actuellement des traitements qui contribue à stabiliser la protéine TTR afin de ralentir sa décomposition et de réduire le dépôt de fibrilles mal repliées.
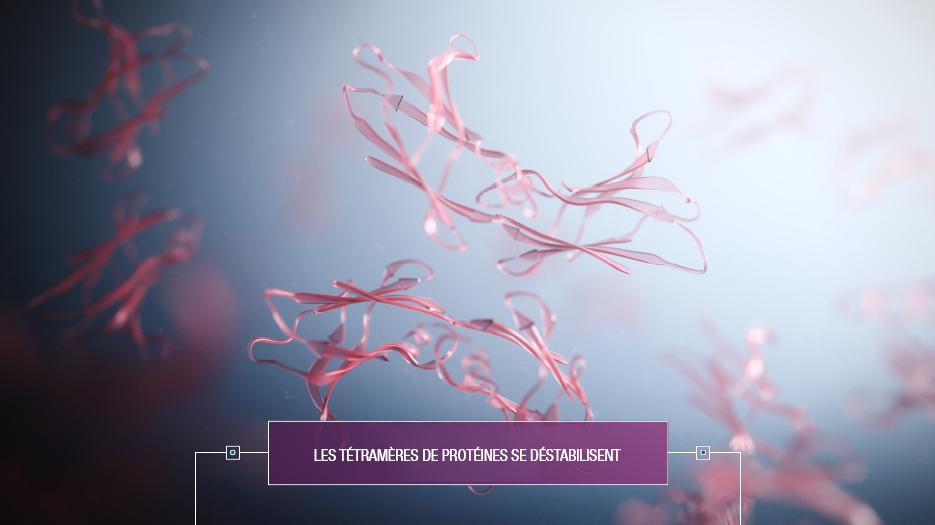

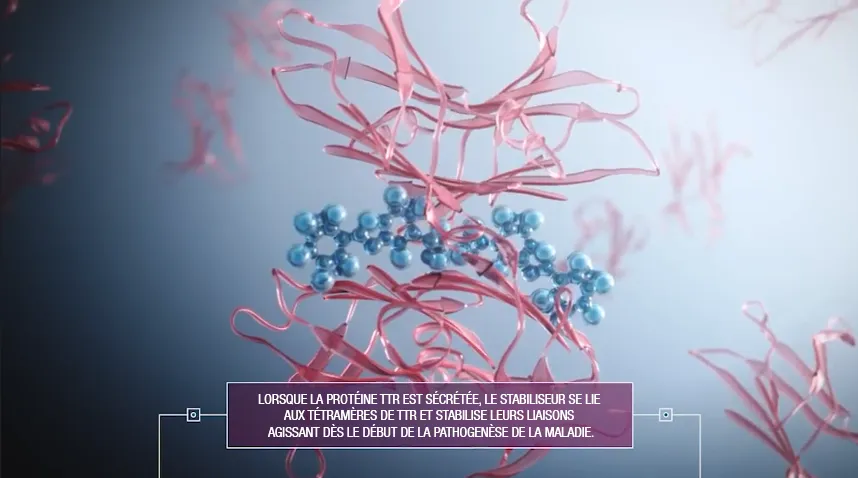
Parallèlement, il existe également des traitements pour soutenir le coeur. Il est aussi possible d'ajouter des traitements pour atténuer les symptômes gênants (diurétique, Beta-bloquants, anticoagulants,..).
D’autres médicaments ayant un mécanisme d’action différent sont encore à l’étude.
Voici quelques astuces utiles :
▶ Prenez toujours vos médicaments au même moment de la journée et veillez à prendre la dose correcte.
▶ Certains patients trouvent pratique d’utiliser un pilulier. Cette boîte peut être remplie une fois par semaine et utilisée comme rappel lorsque vous n’êtes pas sûr d’avoir pris votre médicament ce jour-là. Les piluliers sont disponibles en pharmacie.
▶ N’arrêtez jamais la prise d’un des médicaments prescrits sans consulter votre médecin. Vous risqueriez d’aggraver votre état.
▶ En cas d’hospitalisation, nous vous conseillons d’apporter vos médicaments dans leur conditionnement d’origine.
Soyez particulièrement vigilant si vous présentez les symptômes suivants :
▶ Si vous remarquez une augmentation de l’essoufflement, de la fatigue, du gonflement de l’abdomen ou des jambes, cela peut signifier que vous faites de la rétention d’eau. Vérifiez votre poids et assurez-vous que vous ne consommez pas trop de sel et que vous ne buvez pas trop de liquides. Si les symptômes sont graves ou persistants, n’hésitez pas à contacter l’équipe médicale.
▶ En cas de palpitations ou d’évanouissement, contactez l’équipe médicale.
▶ La faiblesse, les vertiges et les évanouissements peuvent être des signes d’hypotension. Si vous ressentez quelque chose de semblable, prenez votre pouls et votre tension, et contactez l’équipe médicale.
Pour améliorer votre qualité de vie et réduire les symptômes gênants, le médecin peut vous recommander :
▶ Une intervention chirurgicale sur les canaux carpiens et/ou les articulations digitales afin d’éliminer les dépôts de fibrilles amyloïdes
▶ une opération du canal lombaire (en fonction de votre situation cardiaque)
▶ des médicaments comme des laxatifs pour traiter les problèmes digestifs
▶ la prise d’antalgiques
▶ le port d’une prothèse auditive pour corriger un problème d'audition.
Médicaments proscrits :
Les médicaments suivants peuvent aggraver votre fonction cardiaque et/ou rénale :
▶ anti-inflammatoires non stéroïdiens :
antidouleurs comme l’ibuprofène (Brufen® ou Nurofen®), diclofénac, naproxène, piroxicam.
N’utilisez pas ces médicaments sans avoir consulté votre cardiologue !
Antidouleur autorisé :
▶Paracétamol. N’utilisez cependant PAS de comprimés effervescents, car ils contiennent du sel.
S’il n’y a pas d’amélioration au niveau de la douleur, contactez votre médecin ou l’infirmière spécialisée en insuffisance cardiaque.
Références :
1. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy.
N Engl J Med. 2018;379(11):1007–1016. DOI:
10.1056/NEJMoa1805689.
2. Oerlemans M. Cardiac amyloidosis: the need for early diagnosis.
N Engl J Med. 2025;392(15):1234–1242. DOI:10.1056/NEJM20251017001.
3. Nativi-Nicolau, Jose N et al. “Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness.”
Heart failure reviews vol. 27,3 (2022): 785-793. doi:10.1007/s10741-021-10080-2
4. Arbelo E. et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(30):2690–2793. doi:10.1093/eurheartj/ehad194
5. Ruberg F. et al. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019 Jun 11;73(22):2872-2891. doi: 10.1016/j.jacc.2019.04.003.
6. Nature Biotechnology. FDA approves second TTR stabilizer for cardiac amyloidosis. Nature Biotechnology. 2024. Available from:
https://www.nature.com/articles/d41573-024-00188-z. Accessed: October 2025
7. Cleveland Clinic. Amyloidosis (ATTR). Available from:
https://my.clevelandclinic.org/health/diseases/17855-amyloidosis-attr. Accessed: October 2025


