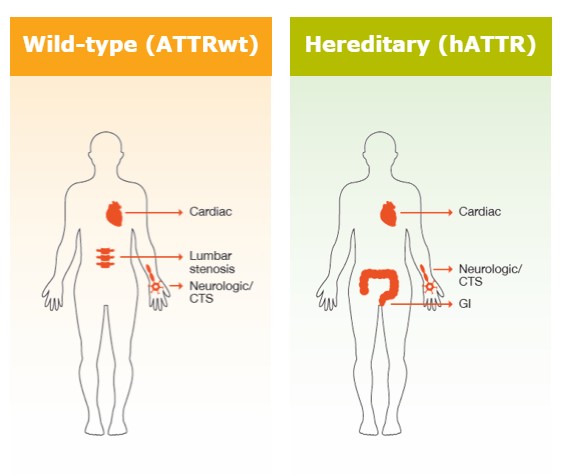
Er zijn twee verschillende vormen van ATTR-CM: wild-type ATTR-CM en erfelijke ATTR-CM8. Elk hebben ze een andere oorzaak.
Wild-type ATTR-CM (wtATTR-CM) komt voornamelijk voor bij patiënten ouder dan 60 jaar; daarom spreekt men ook van ‘cardiale ouderdoms-amyloïdose’. Mannen zijn vatbaarder dan vrouwen. Wat de oorzaak van deze aandoening betreft, tast men nog in het duister. 3,9,10
Daarnaast is er de erfelijke vorm van ATTR-CM (hATTR-CM). Patiënten met hATTR kunnen relatief jong zijn wanneer de symptomen voor het eerst verschijnen, en de evolutie van de ziekte kan van patiënt tot patiënt sterk variëren. Deze vorm komt slechts zelden voor, en treft voornamelijk mannen.9,21
In tegenstelling tot wild-type, is de oorzaak van erfelijke ATTR-CM wél bekend. Patiënten met hATTR-CM hebben namelijk bepaalde veranderingen, of mutaties, in hun genetisch materiaal. Door deze mutaties kunnen er minder stabiele vormen van het transthyretine-eiwit ontstaan. Wereldwijd zijn er meer dan 100 mutaties bekend die in verband gebracht worden met erfelijke ATTR. Afhankelijk van het soort mutatie zal de aandoening een specifiek verloop kennen.9,21
Aangezien ouders genetische mutaties aan hun nakomelingen kunnen doorgeven, spreekt men hier van een erfelijke vorm, ‘hereditary’ in het Engels, vandaar de ‘h’ in de benaming hATTR-CM.9

OVER ATTR-CM
Ons lichaam maakt voortdurend eiwitten aan. Zij vormen de bouwstenen van alle cellen. Deze eiwitten vouwen zich in een bepaalde vorm.
Bij ATTR-amyloïdose – voluit: transthyretine-amyloïdose – loopt er iets mis met de wijze waarop het eiwit transthyretine wordt opgevouwen. De ziekte wordt daarom ook wel de ‘eiwitvouwziekte’ genoemd. Door het verkeerd opvouwen, ontstaan er ‘onstabiele’ eiwitten die samenklitten. De eiwitketens die zo ontstaan noemt men ‘amyloïdfibrillen’. 1, 2 Je lichaam kan deze fibrillen niet meer verwerken of uitscheiden, waardoor ze neerslaan in organen of weefsels, en daar schade veroorzaken.
Afhankelijk van de plaats waar de fibrillen in het lichaam worden afgezet, kan de ziekte op diverse wijzen tot uiting komen. Gebeurt dit in het hart, dan krijg je hartklachten; in de nieren veroorzaken ze nierfalen; in het zenuwstelsel leiden ze tot chronische pijn of gevoelsstoornissen. 3,4
Zonder behandeling hebben patiënten met ATTR-amyloïdose een gemiddelde levensverwachting van 3 tot 5 jaar vanaf het moment van diagnose. Met een gepaste behandeling kan je het ziekteproces afremmen en de levensverwachting verlengen.5
ATTR-CM staat voor transthyretine-amyloïdose cardiomyopathie (CM)6,7. Bij deze vorm van amyloïdose zetten de amyloïdfibrillen zich af in de hartventrikels of hartkamers. Dit zijn de holten in het hart die er door samentrekking van de hartspier voor zorgen dat het bloed in de slagaders gestuwd wordt.
Wanneer de amyloïdfibrillen zich hier afzetten, worden de spieren van de hartventrikels dikker en verstijven ze. Dat leidt uiteindelijk tot een minder goede pompwerking van het hart, waardoor je zuurstofrijk bloed niet meer voldoende kan worden rondgepompt en je ernstige hartklachten kan krijgen.8

Wanneer er op basis van bepaalde symptomen een vermoeden is van ATTR-CM, kunnen gespecialiseerde testen de ziekte bevestigen. De belangrijkste testen zijn een echocardiografie (ECG), aangevuld met geavanceerde beeldvormingstechnieken (zoals een MRI en botscan), laboratoriumonderzoek en genetische testen10,11.
Bij patiënten die lijden aan de erfelijke vorm, kan het nuttig zijn om de naaste familieleden eveneens te testen. Familieleden die positief testen, kunnen immers nauwgezet worden opgevolgd en een gepaste behandeling krijgen van zodra er symptomen opduiken9.
Ook na de diagnose blijft het voor de patiënt belangrijk om geregeld onderzoeken te ondergaan – onder meer echocardiografie en bloedanalyse – om de progressie van de ziekte op te volgen.
Je arts bepaalt welke onderzoeken voor jou zinvol zijn en zal je hierover informeren.
Het ziekteverloop verschilt van patiënt tot patiënt, en is afhankelijk van je algemene conditie, je leeftijd en vooral ook het stadium waarin de ziekte wordt vastgesteld.
Een vroegtijdige diagnose is belangrijk om de ziekte zo goed mogelijk te stabiliseren en het ziekteverloop te vertragen. Zodra je een diagnose hebt, kan je immers de behandeling opstarten en de toename van de hoeveelheid amyloïdfibrillen in het lichaam afremmen. Dit zorgt voor een betere levenskwaliteit en een verlenging van de levensverwachting.
Maar omdat patiënten erg uiteenlopende symptomen kunnen hebben, zowel cardiale als niet-cardiale, klachten die bovendien weinig specifiek zijn, is het niet altijd eenvoudig om een diagnose te stellen in een vroeg stadium. Bij een laattijdige diagnose is de prognose minder gunstig


De symptomen zijn afhankelijk van de plaats in het lichaam waar amyloïd wordt afgezet. Aangezien het bij ATTR-CM gaat om een aandoening van de hartspier, treden hierbij vooral hartproblemen op. De klachten lijken sterk op die van hartfalen, maar kunnen van patiënt tot patiënt verschillen. 14-16
Symptomen |
Klachten |
|---|---|
Hartfalen |
• Kortademigheid (dyspneu) |
Andere vaak voorkomende symptomen zijn: 2, 16, 17
Symptomen |
Klachten |
|---|---|
Polyneuropathie |
• Gevoelsstoornissen/pijn op verschillende plaatsen in het lichaam door aantasting van het perifere zenuwstelsel |
(Bilateraal) Carpaaltunnelsyndroom |
• Dof, tintelend of pijnlijk gevoel in de hand(en) en/of vingers door een knelling van de zenuw ter hoogte van de pols(en) |
Wervelkanaalstenose |
• Pijnlijke, vermoeide benen (zwaar gevoel) |
Naast het hart kan ook het zenuwstelsel worden aangetast. Dit leidt tot polyneuropathie, een aandoening van het perifeer zenuwstelsel.
Sommige patiënten krijgen ook te maken met carpaaltunnelsyndroom, dat is compressie van de zenuw in het polskanaal. Omdat dit syndroom opduikt in een vroeg stadium van het ziekteproces voordat enig ander detecteerbaar symptoom ontstaat, kan dit een vroeg teken van ATTR-CM zijn, maar is dat niet altijd.
Aangezien het ziektebeeld sterk kan verschillen van patiënt tot patiënt, is het mogelijk dat er nog andere bijkomende, niet-cardiale klachten optreden. Zo kan je ook volgende symptomen hebben: 16, 18
Symptomen |
Klachten |
|---|---|
Maagdarmsymptomen |
• Diarree |
Intolerantie voor geneesmiddelen die hartfalen behandelen11, 17, 19 |
- |
Scheur van de bicepspees |
- |
Heup- of knieproblemen met prothesen als gevolg18 |
- |
Oogaandoeningen4 |
• Druk op de ogen |
Gezien de weinig specifieke aard van deze klachten, is het niet altijd evident om de juiste diagnose te stellen.3,4

Deze illustratie geeft een aantal mogelijke symptomen en klachten weer. Deze zijn erg uiteenlopend en kunnen variëren van patiënt tot patiënt.
Cardiale amyloïdose is vandaag nog niet te genezen. Wel is de ziekte steeds beter behandelbaar met geneesmiddelen die het ziekteproces kunnen afremmen4. Je arts kan je hier zeker meer over vertellen.
Naargelang de specifieke symptomen die je hebt, zal je arts ook medicijnen voorschrijven om de hinderlijke klachten te verminderen. Plaspillen bijvoorbeeld, of medicatie voor de behandeling van hartfalen; eventuele pijn kan je bestrijden met pijnstillers.
Omdat het de lever is die het instabiele transthyretine-eiwit produceert, kan in sommige gevallen een levertransplantatie overwogen worden. Deze ingreep is echter niet voor iedereen mogelijk en brengt risico’s met zich mee. Als je hiervoor in aanmerking komt, zal je arts je hierover ongetwijfeld informeren.
Besteed tot slot ook aandacht aan je levensstijl. Een aangepaste voeding en voldoende beweging bijvoorbeeld kunnen een gunstige invloed hebben op je algemene levenskwaliteit. Vraag hierover advies aan je arts.13,20
1.Siddiqi OK, Ruberg FL. Trends Cardiovasc Med. 2018;28(1):10–21.
2.Halwani O, Delgado DH. Expert Rev Cardiovasc Ther. 2010;8(7):1007–1113.
3.Donnelly JP, Hanna M. Cardiac amyloidosis: an update on diagnosis and treatment. Cleve Clin J Med. 2017;84(12 suppl 3):12-26.
4.Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, Gospodinova M, Obici L, Rapezzi C, Garcia-Pavia P. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019 Aug;7(8):709-716. doi: 10.1016/j.jchf.2019.04.010. Epub 2019 Jul 10. PMID: 31302046.
5.Ruberg, F.L., Grogan, M., Hanna, M., Kelly, J.W., Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol, 2019, 73 (22), 2872-2891.
6.González-López E, López-Sainz Á, Garcia-Pavia P. Diagnosis and treatment of transthyretin cardiac amyloidosis. Progress and hope. Rev Esp Cardiol. 2017;70(11):991-1004.
7.Quarta CC, Solomon D, Uraizee I et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129(18):1840-1849.
8.Maurer MS, Hanna M, Grogan M et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol,. 2016;68(2):161-172.
9.Oerlemans MIFJ, Rutten KHG, Minnema MC, Raymakers RAP, Asselbergs FW, de Jonge N. Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 2019;27(11):525-536.
10.Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.
11. Narotsky D, et al. Wild-type transthyretin cardiac amyloidosis: novel insights from advanced imaging. Can J Cardiol. 2016;32(9):1166.e1-1166.e10
12. TTR-FAP: A Quick Guide for General Practitioners. Pfizer 2017.
13. Adams D. et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016, 29 (suppl 1):S14–S26.
14. Mayo Clinic: Heart failure. http://www.mayoclinic.org/diseases-conditions/heart-failure/symptoms-causes/syc-20373142. Accessed May 8, 2019.
15. Ponikowski P, et al. Eur Heart J. 2016;37(27):2129–2200.
16. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, Grogan M, Kristen AV, Lousada I, Nativi-Nicolau J, Cristina Quarta C, Rapezzi C, Ruberg FL, Witteles R, Merlini G. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019 Sep;12(9):e006075. doi: 10.1161/CIRCHEARTFAILURE.119.006075. Epub 2019 Sep 4. PMID: 31480867; PMCID: PMC6736650.
17. Brunjes DL, Castano A, Clemons A, Rubin J, Maurer MS. Transthyretin cardiac amyloidosis in older Americans. J Card Fail. 2016;22(12):996–1003.
18. Rubin J, Alvarez J, Teruya S, Castano A, Lehman RA, Weidenbaum M, Geller JA, Helmke S, Maurer MS. Hip and knee arthroplasty are common among patients with transthyretin cardiac amyloidosis, occurring years before cardiac amyloid diagnosis: can we identify affected patients earlier? Amyloid. 2017 Dec;24(4):226-230. doi: 10.1080/13506129.2017.1375908. Epub 2017 Sep 14. PMID: 28906148.
19. Castaño A, Drach BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-178.
20. Pinney JH & Hawkins PN. Amyloidosis. Review Article. Ann Clin Biochem 2012; 49: 229–241. DOI: 10.1258/acb.2011.011225.
21. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016;68(12):1323-41


